![]() Phosphate Induced Metal Stabilization (PIMS)
Phosphate Induced Metal Stabilization (PIMS)
Apatite remediation of heavy metals is an emerging technology that addresses the need to remediate metals in wastes, contaminated groundwater, sediments and soils, including agricultural soils. Metals readily leach from contaminated soils, sediments and particular fertilizers (especially some phosphate and zinc-enriched formulations) and serve as a constant source of metal contamination to surface waters, underlying groundwater zones, and ingestion pathways of the biota. Estimates of remedial costs for mining operations, agricultural lands, industrial manufacturing, landfills and national laboratories have exceeded one hundred billion dollars. Efforts to mobilize and remove metals from the subsurface to below regulatory limits have not been successful because of the various intermediate solubilities and sorption properties that each metal and suite of metals exhibits under most environmental conditions. Total removal of the contaminated material for disposal elsewhere is not feasible, as it exceeds all landfill space presently available in the United States. Alternately, metals can be stabilized in place to prevent them from migrating or leaching into groundwater or the ecosystem. Many materials have been proposed for this purpose, but the technology described here is particularly effective for non-redox-sensitive metals for which no adequate, cost-effective alternatives exist, e.g., lead, cadmium and zinc. This technology stabilizes metals by chemically binding them into new stable phosphate phases (apatite minerals) and other relatively insoluble phases in the soil, sediment or in a permeable reactive groundwater barrier. Metals most effectively stabilized by this treatment are lead, zinc, copper. cadmium, nickel, uranium, barium, cesium, strontium, plutonium, thorium, and other lanthanides and actinides. Because of its extreme toxicity, lead has been the focus of these studies.The mineral apatite is necessary for this technology. Non-apatite phosphate and mixtures of precursor constituents will not perform as well, and usually not at all. The apatite works in several ways. Specific metals, such as lead, can enter the apatite mineral structure during precipitation of new solids, e.g., lead-apatite, or by exchanging with calcium in an existing calcium-apatite. Nucleation on existing apatite is important. Apatite is also the best natural material available for non-specific adsorption. In addition, apatite is an excellent buffer for neutralizing acidity through P043-, OH-, and substituted CO32-, and exerts control on the chemical activities of other species leading to the precipitation of oxide-, hydroxide- and carbonate-metal phases.
Metals sequestered in apatite minerals have great durability and leach resistance significantly exceeding other chemically stabilized forms. This is because the apatite mineral structure is very stable over a wide range of environmental conditions, e.g., pH 2 to 12, up to 1000 degrees C, in the presence of aqueous and non-aqueous phase liquids, and under disruptions such as earthquakes, ground subsidence or human intrusion, for geologically long time periods, i.e., hundreds of millions of years. Therefore, the metal-enriched apatites will no longer be a source for further groundwater contamination. Also, because of the long-term stability of metal-enriched apatites (Altschuler et al., 1967; Kovach and Zartman, 1981; Shaw and Wasserburg, 1985; Keto and Jacobsen, 1987), the effects of gravity, soil heterogeneity, hydrology and other properties of the subsurface do not affect the performance of the remediation treatment using apatites, and the subsurface conditions are not affected by this treatment. The bioavailability of ingested metal- apatite is also greatly reduced (Davis et al, 1992; Ruby et al, 1992), making animal and human intrusion less dangerous should the metal-apatite phase be ingested, and making bioremediation more effective in mixed waste environments. The reaction between the apatite and metals is rapid (Ma et al., 1993; Wright et al., 1995; Chen et al., 1997a,b), and so the treatment is effective immediately, requiring no time for the material to "set up. " Previous results indicate that as little as 1% by weight of apatite could remediate most metal-contaminated soils, even leaching lead-acid battery sites, avoiding volume problems associated with many other methods. For groundwater, a permeable reactive barrier of apatite can immobilize over 20% of its weight in metals, and becomes an ore with separate economic value. For contaminated agricultural soils, the apatite immobilizes metals already present from previous activities, and provides phosphate to the soil in appropriate amounts for long-term fertigation. Apatite II can also be adapted to provide zinc to the soil without the other unwanted metals associated with the present micronutrient blends, granular zinc blends and phosphate fertilizers (Bowhay et al., 1997).
There are several apatite sources with widely varying reactivities and properties, and not all are appropriate for metal remediation. For metal remediation, the apatite should 1) be fully carbonated with as much carbonate ion substituted as possible, 2) have no fluorine substitution in the hydroxyl position, 3) have little or no trace metals initially in the structure, and 4) have a high internal porosity. For these reasons, traditional phosphate ores are not useful. The Apatite II developed for this purpose at UFA Ventures exhibits all of these properties, is the most effective and the least expensive apatite available, and costs approximately $500/ton, including transportation.
Other processes and remediation technologies for other contaminants are not inhibited by apatite treatment, e.g., agricultural uses will not compromise this treatment, and metal-stabilization by apatite treatment will not affect vapor stripping or bioremediation of organics from the same soils in a mixed-waste system. For applications in which both chemical stabilization and physical solidification are desired (in landfills, for example), apatite can be combined with grout for improved stabilization performance. Apatite is also ideal as an additive to disposal facility liners, and can be used with other additives, such as bentonite or zeolite. Finally, this treatment is possible using existing emplacement technologies, e.g., auguring into contaminated soils, slurry injection, excavation and backfill, or above-ground treatment of contaminated groundwater. This technology is fully commercialized.
The results of previous and ongoing work demonstrate that stabilization of contaminated soils and groundwater by the mineral apatite has the potential to be the most successful and widely applicable remediation strategy for metals and radionuclides in the foreseeable future. Utilizing apatite minerals as a remediation method puts to use long-recognized geochemical principles. The groundwork for this research has been laid by previous studies in widely divergent disciplines, including 1) phosphate mineralogy and crystal chemistry (Skinner, 1987, 1989; Skinner and Burnharn,1968; Wright,1990a,b; Wright et al., 1990); 2) scavenging and sequestration of minor and trace elements, such as uranium, metals, and the rare earth elements, in natural phosphate deposits (Altschuler et al., 1967; Kovach and Zartman, 1981; Shaw and Wasserburg, 1985; Keto and Jacobsen, 1987; McArthur et al, 1990); 3) remediation studies of phosphate/lead systems (Chen et al., 1997a,b; Ruby et al., 1994; Xu and Schwartz, 1994; Stanforth and Chowdhury, 1994; many papers from T. Logan's group, especially Ma et al., 1993); 4) the impact and accessibility of phosphorus fertilizers to crops (Adepoju et al, 1986); 5) natural analogues in metallic mineral deposits (Koeppenkastrop and DeCarlo, 1988, 1990); 6) phosphate diagenesis during the formation and evolution of phosphorite deposits (McArthur, 1985) and 7) the evidence of changes in the paleochemical evolution of oceans, atmospheres, and climates evidenced by metals, lanthanides, and actinides incorporated into fossil teeth that have an apatite composition (Shaw and Wasserburg, 1985; Keto and Jacobsen, 1987; Wright, 1990a,b).
Apatite minerals form naturally and are stable across a wide range of geologic conditions for hundreds of millions of years (Nriagu, 1974; Wright 1990). Work by Wright, Conca and others (Kovach and Zartman, 1981; Shaw and Wasserburg, 1985; Keto and Jacobsen, 1987; Wright et al., 1987a,b, 1984, and 1990) investigated the trace element composition of apatite in fossil teeth and bones and in sedimentary phosphorite deposits through geologic time. They found that sedimentary and biogenic apatite deposited in seawater concentrates metals and radionuclides from the seawater to millions of times the ambient concentration, and locks them into the apatite structure for up to a billion years with no subsequent desorption, leaching or exchange, even in the face of subsequent diagenetic changes in the pore water chemistry, pH, temperatures up to 1000 degrees C, and geologic or tectonic disruptions, e.g., uplift, subsidence, erosion and earthquakes. Over 300 apatite minerals exist, with elements from the entire periodic table replacing calcium, phosphate, and hydroxide in the fundamental apatite crystal structure (Deer et al., 1978; Skinner, 1987, 1989).
The ultimate driving force for the potentially robust performance of reactive phosphate with respect to metals is the extreme stability of these metal-phosphate phases, some of which are shown in Table 1. The solubilities of quartz and common table salt are also shown. Common table salt is normally considered as being very soluble and quartz is normally considered as being fairly insoluble. However, these metal phosphates are twenty to seventy orders of magnitude more insoluble than quartz. Combined with this thermodynamic stability, the rapid kinetics of the metal-phosphate precipitation ensures immobilization of the metals in the face of most possible transport mechanisms.
TABLE 1. Solubilities of Some Metal-Phosphate Phases.
Mineral Phase Solubility Product
(log Ksp)Pb5(P04)3(OH,Cl) -76.5 Sr5(P04)3(OH) -51.3 Zn3(P04)2 -35.3 Cd3(P04)2 -32.6 Am(P04) -24.8 Pu(P04) -24.4 Salt (NaCl) 0.0 Quartz (SiO2) -4.0 Our research group recently investigated the metal-stabilization potential of reactive phosphates from different sources under grants from the Strategic Environmental Research and Development Program (SERDP, Department of Defense), EPA and Idaho State Department of Environmental Quality (Chen et al., 1997a,b; Wright et al., 1995). Our preliminary studies used:
1) soils containing 1000-4000 mg kg-1 (ppm), or 0.1-0.4% by weight, of lead (Pb), as well as high concentrations of zinc (Zn) and cadmium (Cd), and
2) groundwaters contaminated with Zn, Pb, Cd and Cu up to concentrations of 250 ppm, 10 ppm, 1 ppm and 20 ppm, respectively.
Data from the chemical and mineralogical speciation studies were used in the geochemical code MINTEQ-A2 to determine the stability relationships of the soil and metal-apatite minerals under varying conditions and the metal-apatite phases were predicted to be the most stable under many expected subsurface conditions.
Batch and column tests are the two types of laboratory experiments usually performed to determine performance in sorption systems. In the desorption or leaching batch studies, water is added to samples in centrifuge tubes. The water to soil ratio of 10:1 is selected according to established testing protocols and is useful for studying the basic thermodynamics of the system. The concentration of contaminants in solution is monitored over time for up to 336 hours. In the flow- through leaching column tests, samples are packed into columns. Water is introduced at the top of the sample, and leachates are collected as a function of the number of pore volumes of solution added, or as a function of the mass of sorptive material which reflects a reactive barrier application. These experiments are performed with the actual systems, e.g., contaminated soil or groundwater as collected from the field, and with the soil treated with various added amounts of apatite mineral. Results from these recent studies showed that the leachates from untreated soils were in the hundreds to thousands of mg kg-1 (ppm) range for Pb and Zn, well in excess of Maximum Contamination Levels (MCLs) for drinking water. Cd was hundreds of mg kg-' (ppm), also in excess of the MCL for drinking water. However, leachates from apatite-treated soils showed concentrations for Pb, Zn and Cd below the detection limits of 1 µg kg-' (ppb) using Inductively-Coupled Plasma Mass Spectrometry (ICP/MS) or Potentiometric Stripping.
Metal-Apatite Adsorption Isotherms and Batch Tests - Top of page
Adsorption is most often described in terms of isotherms which show the relationship between the bulk activity, or effective concentration in solution, of the species being adsorbed and the actual amount adsorbed at a constant temperature. When plotted, the shape and mathematical expression of the isotherm provide substantial information concerning the chemistry and sorption mechanisms within the system.
Although current research demonstrates the successful precipitation of soluble heavy metals when hydroxyapatites are added to a contaminated medium in batch studies (Misra and Bowen, 1981; Ma, et al. 1993; Xu and Schwartz, 1994), very little attention has been paid to performance in actual soil systems or to the precise amount of hydroxyapatite needed to affect precipitation or complexation of metals from contaminated mediums. In our past work, the amounts of hydroxyapatite needed to precipitate soluble metals relative to the molar ratios of the precipitated metal-P04 complexes are illustrated as sharp deviations from linearity of the adsorption isotherms, as predicted by MINTEQ-A2, a thermodynamic speciation model (Moody and Wright, 1995; Wight et al., 1995). The amounts of apatite required for precipitating the metals in solution can then be determined for field remediation. As an example, two-week desorption experiments on untreated soils from the Bunker Hill Mining Site resulted in soluble Pb, Zn, Cd and Mn concentrations which are presented in Figure 1. Pb, Zn and Cd are of regulatory concern. Note that the maximum amount of metal desorption occurs within the first 24 hours of desorption. The concentrations of metals in the leachates are from tens to tens of thousands of µg kg-' (ppb) and establish the general ranges of metal concentrations to be expected during leaching and which must be treated.
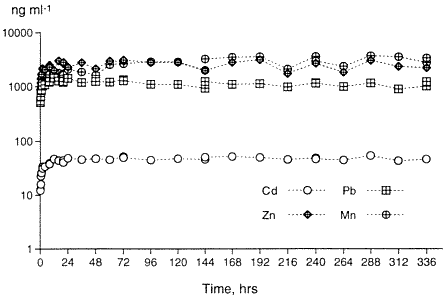
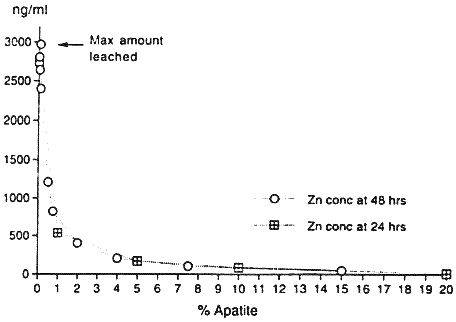
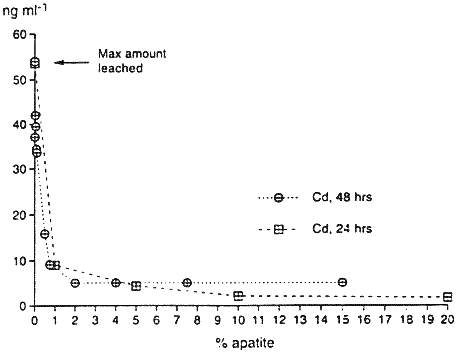
Figure 1. Desorption of Pb, Zn, Mn and Cd From the Contaminated Soil During a Two Week Period.Adsorption isotherm tests (Beckwith, 1964) using sedimentary and biogenic apatites, were also performed on the contaminated soils. For 24-hour and 48-hour apatite adsorption studies, 3 grams of the contaminated soil, ground to pass through a 170 mesh screen and weighed to three decimal places, were combined by simple mixing with various amounts of apatite from 0.005% by weight to 20% by weight. Thirty milliliters of deionized water were added to the mixture in 40 ml polycarbonate centrifuge tubes. The polycarbonate tubes containing the 3 grams of contaminated soil, incremental percentages of apatite and 30 ml of deionized water were shaken continuously for 24 hours and 48 hours in separate experiments. At the end of these times, the samples were taken from the shaker and centrifuged. The supernatant was filtered at 0.2µ, then analyzed for pH, metals, and anions. The results are shown in Figure 2. At 0% apatite (the control sample), the desorbed amount of Pb, Zn and Cd is 1600 ng ml-1, 2760 ng ml -1, and 54 ng ml -1, respectively. For the apatite-treated samples, the greatest reduction occurs with only 1% addition of apatite. Increasing the adsorption isotherm time from 24 hours to 48 hours did not increase the overall effectiveness of apatite, suggesting that the adsorption or formation kinetics of the apatite-metal system for these metals is less than 24 hours. These soil studies corroborate the batch studies of other researchers in metal-P04 complexation (Ma, et al, 1993, Ruby, et al, 1994, Xu and Schwartz, 1994).
Figure 2. The Reduction of Desorbed Pb, Zn, and Cd by NC Apatite Depicted by 24 and 48 Hour Adsorption Isotherms for Bunker Hill Contaminated Soil.
Model Verification of Precipitated Metals using MINTEQ-A2 - Top of page
In order to gain knowledge about the nature of metal-phosphate complexation and to determine how much P04 is needed to complex the desorbed metals of the contaminated soil, MINTEQ-A2, a geochemical thermodynamic speciation computer program, was used. The natural soil system has both a solid phase and a solution phase. When chemical equilibrium is assumed to exist between the soil solution and the associated solid phases of the soil, one can elicit important information about solid phase formation by using thermodynamic calculations performed in geochemical thermodynamic speciation programs. Specifically, this computer program is used to examine the precipitation of selected metals, i.e., Pb, Zn, and Cd, induced by the application of a specific phosphate compound. Saturation indices for the contaminated soils determined from MINTEQ-A2 indicated that Pb, Zn, Mn, and Cd-phosphate minerals are the most stable phases in these systems under subsurface conditions. The saturation index is defined as the Ion Activity Product divided by the Solubility Product and is a thermodynamic indication of mineral dissolution or formation. The higher the saturation index, the greater the probability of precipitation of that mineral. Figure 3 shows the MINTEQ-A2 results for saturation indexes relative to apatite concentrations for the formations of Pb-phosphate minerals. The Pb-P04 minerals depicted in Figure 3 are all common minerals known to form under the given conditions. The chloro- and hydroxypyromorphytes, with the chemical formula Pb 5(P04)3(OH, Cl), are indicated as having the most positive saturation indexes, and are, therefore, most likely to form. This was substantiated by X-ray diffraction results on the contaminated soils after treatment with apatite (Wright et al., 1995) and is also consistent with results by other researchers (Nriagu, 1974, Ma, et al, 1993, Ruby et at, 1994). Plumbogummite (PbAl3(P04)2(OH)5 H20) is also indicated as a possible precipitated mineral, as are two unnamed complexes, Pb(P04) and PbHPO4. For all of the Pb-P04 minerals presented in the MINTEQ-A2 output, the thermodynamic predictions occur at less than 0.1% addition of apatite (Moody and Wright, 1995). This thermodynamic prediction agrees with the batch test results shown in Figure 2. Similar results were obtained for Zn and Cd. The metal-apatite phases are the most stable for these metals, and for most other metals of interest, e.g., chromium, copper, nickel, uranium, barium, cesium, strontium, plutonium, thorium, and the other lanthanides and actinides.
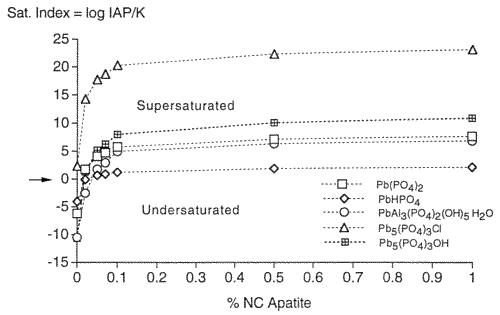
Figure 3. Saturation Indexes Relative to Increasing Amounts of Apatite for the Formation of Pb-Phosphate Minerals.
Metal-Apatite Adsorption in Column Tests under Subsurface Conditions - Top of page
Flow-through studies of untreated and apatite-treated contaminated soils were performed to replicate actual field conditions that batch studies cannot reproduce, e.g., natural flow rates, low water:soil ratios, channelized flow paths, etc. Two flow methods were used: traditional soil columns and the Unsaturated Flow Apparatus (UFATM) method. Traditional soil columns consist of a column of soil having solution dripping into the top at a fixed rate and effluent collected as it exits the bottom of the sample. The degree of saturation in the sample is high, nearly saturated, for most soils if the experiment is to take less than a year. The UFA method is used to achieve the low water contents that exist in most vadose zones and for flow in relatively impermeable materials (Conca and Wright, 1998). Two 3-cm diameter, 5-cm long core sample holders were each packed with 40 g of dry contaminated soils for the UFA runs and for the traditional soil column experiments. Cores of the contaminated soils were run as untreated cores and as treated cores. Treated cores were treated with North Carolina natural apatite in two different ways, either as a layer at the bottom of a UFA sample holder to reproduce the effect of field-emplaced permeable reactive barrier or simply mixed into the soils as an additive to reflect auguring or soil mixing as a field emplacement technology. Leachates were analyzed by ICP/MS on both unfiltered portions and portions filtered to 0.45 µm. Filtering had no effect on the results.
Figure 4 shows some results. In all experiments, regardless of how the apatite was emplaced, no metals were leached from the apatite treated soils above the ICP/MS detection limits of I ppb for Pb and Zn, and 5 ppb for Cd. On the other hand, high concentrations of metals were leached from the untreated soils in all cases. More than any other work, these column results on real, highly- contaminated soils indicate the great potential of this method for remediating metal-contaminated soils and groundwater.

Figure 4. Soil Column Test Desorption/Sorption Run on Bunker Hill Contaminated Soil with 1 percent Apatite by Weight Simply Mixed (as with Emplacement by Augering into Contaminated Soil) using Vadose Zone Water at a Recharge of 2 x 10-7 cm/s.
Success Mine Project - Top of page
In preparation for emplacement of a permeable reactive barrier to treat Zn, Cd and Pb-contaminated seep waters exiting the base of the Success Mine tailings pile in northern Idaho, the Idaho State Department of Environmental Quality commissioned our laboratory to run a feasibility study of the various materials that might be used, including apatite. We investigated four different sources ot apatite (three Apatite II formulations [Apatite WE, PR, and AP] and another type of apatite [Apatite CB]), iron-filings that work for remediating chromium and other metals, compost/wood chip/gravel mixtures similar to that used in the Canadian work, two zeolites and a polymer used in remediation of mine wastes, and activated charcoal. In all of the column tests, only Compost showed breakthrough of all four metals, including Cu and Pb, and it occurred quite rapidly. Overall, Apatite WE performed the best with respect to all metals. Column tests used polypropylene columns with micron-filter ends and flow rates of 3 ml/hr giving average water residence times of about 1 hour. The experiments were stopped for columns that showed rapid breakthrough for Zn. Pb and Cu did not break through any columns except for the Compost. The results for Zn are shown in Figures 5 and 6 which are plots of contaminant concentration in the effluent normalized to the influent, C/C0, versus the volume of water passing through the column normalized to the weight of barrier material. This normalizes the columns so that they can be compared on a weight basis because these materials are purchased and emplaced on a weight basis, not a volume basis. Breakthrough is usually taken to be when C/C0=0.5. However, significant removal can still occur after this point if the slope of the breakthrough curve is low, as with the apatites.
The Iron Filings failed within 24 hours with respect to Cd. The Zn shows a linear breakthrough in the Iron Filings that began almost immediately, indicating that it is inappropriate for this type of remediation. Also, the Iron Filings produced significant colloids that travelled through the column and exited with the effluent. Zn, Cd, Cu and Pb began to breakthrough the Compost at the ppm (Zn) and ppb levels (Cd, Cu, Pb) after only 10 pore volumes, indicating that Compost is inappropriate for this type of remediation. The Apatite CB shows low level breakthrough with respect to Cd and its Zn performance is not much better than that of the Iron Filings. Apatite PR began to breakthrough with respect to Zn after only 150 pore volumes, but was still removing a significant fraction of Zn from solution even after 500 pore volumes. When the experiments were stopped, Apatite PR was beginning to show incipient breakthrough with respect to Cd, but the other two apatites were still below detection for Cd (< 5 ppb Cd). Even after significant breakthrough, Apatites WE, PR and AP were still removing a very significant fraction of Zn. The clinoptilolite, C-sorb and NC Apatite broke through rapidly. Cabsorb, which is the zeolite chabazite, acted like a perfect cation exchanger and broke through in a step function after 100 pore volumes.
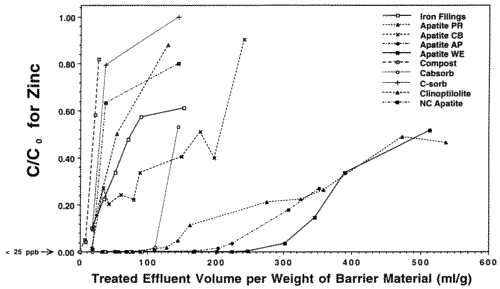
Figure 5. Column Breakthrough Tests with Various Candidate Materials for an In-place Permeable Reactive Barrier at Success Mine. Co = 150 ppm Zn
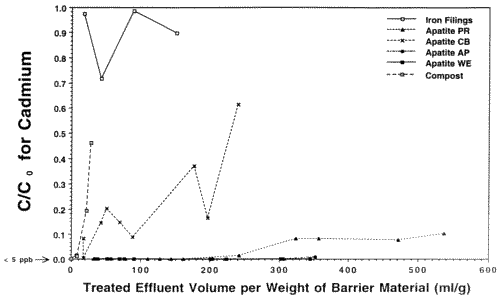
Figure 6. Column Breakthrough Tests with Various Candidate Materials for an In-place Permeable Reactive Barrier at Success Mine. Co = 1 ppm Cd
Different barrier materials buffer the pH to varying degrees. pH was taken on the influent and on the effluent for the columns at the beginning of the experiment and after about 300 pore volumes had exited the columns (P.V.).
Barrier Material Influent pH Effluent pH (at 50 P.V.) Effluent pH (at 300 P.V.) Iron Filings 4.57 4.33 5.10 Clinoptilolite 4.57 4.15 NR Cabsorb 4.57 4.09 NR C-Sorb 4.57 6.57 NR NC Apatite 4.57 6.70 NR Apatite PR 4.57 7.15 6.21 Apatite CB 4.57 7.37 6.50 Apatite AP 4.57 7.00 6.54 Apatite WE 4.57 6.87 6.79 Compost 4.57 6.84 NR NR - not run as these columns failed after 100 P.V. These tabular results should be compared to the breakthrough curves shown in Figures 5 and 6. The apatites and the C-sorb provide significant buffering, while the iron filings and the zeolites do not provide significant buffering. The compost provides intermediate buffering but was only run for 10 pore volumes before breakthrough occurred for the metals.
Ingestion Batch Tests - Top of page
Batch leach tests were performed to provide a relative effect of phosphate on the bioavailability of Pb from ingested contaminated soils in swans, a severe problem in this area. Contaminated soils were collected that were 1) not treated, designated -PO4; 2) treated with a standard amount of phosphate fertilizer, designated +PO 4 and treated with twice the standard amount of phosphate fertilizer, designated ++PO 4; and 3) treated with 1%, 10% and 50% Apatite II by weight, designated 1% apatite, 10% apatite and 50% apatite, respectively. These were run in batch tests with vadose zone water adjusted to pH 2.4 with HCl. The substrate to water ratio was the standard 1:10 and mixtures were shaken for 24 hours. The solutions were then filtered through 0.2 µm filters and analyzed for Pb and Zn. The results are given below.
Soil Pb Zn pH -PO4 24,000 ppb 2,800 ppb 4.28 +PO4 120 ppb 3,800 ppb 4.37 ++PO4 10 ppb 4,000 ppb 5.49 1% apatite 174 ppb 140 ppb 6.07 10% apatite 104 ppb < 50 ppb 6.87 50% apatite 240 ppb < 50 ppb 7.02 These results indicate that the presence of phosphate in either form dramatically reduces the bioavailability of Pb. The apatite also provides good pH buffering in this system and reduces the availability of Zn, which occurs in high concentration in these fertilizers. The fertilizer reduced the Pb concentrations slightly more than the apatite, but fertilizer is highly soluble and will require repeated application in the field. The performance of the apatite seems not to depend upon the amount of apatite used, with the concentration of Pb in solution seeming to be equilibrium controlled at the hundreds of ppb level.
Lead-Loading Experiments - Top of page
Pb never broke through any of the retardation columns. However, because of the toxicity of Pb (MCL= 15 ppb) and its great effect on learning disabilities in children even at low concentrations, it is desirable to know the Pb holding capacity, or retention efficiency, of the apatites. Therefore, a total Pb loading experiment was performed on Apatite II and two ordinary apatites (unground and ground to 170 µm) to determine just how much Pb they can immobilize. A 500 ppm (mg/L) solution of Pb as Pb(NO3)2 was infiltrated into columns containing 20 g of each material at a flow rate of 2 ml/hr. The results are given in Figure 7. The experiments had to be halted because of time constraints, and only the unground apatite column began to show signs of breakthrough but with a C/C0 of only 0.08. As a control, uncontaminated Bunker Hill soil broke through immediately. Each apatite retained more than 17% of its weight in Pb. Theoretical efficiencies are 40%. Because of the high concentration of Pb in the influent, the number of pore volumes passed was only 1200 at the time the experiment was stopped, but at the Pb concentrations of most contaminated waters and leachates, this would be equivalent to tens of thousands of pore volumes, an enormous subsurface flux representing tens of years at normal discharge fluxes in the subsurface. The reason for the extreme efficiency is that Pb is precipitating as pyromorphite (Chen et al., 1997). This efficiency will not be much affected by the presence of competing Zn and Cd (Ma et al., 1994; Wright et al., 1995; Chen et al., 1997), which appear to be both surface sorbing and forming various phases such as otavite, hydrocerussite, zincite and hopeite.
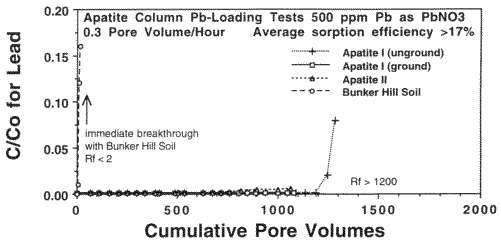
Figure 7. Pb-Loading Column Breakthrough Tests with 500 ppm Pb Influent.
References - Top of pageThe cost savings are tremedous, over 95% savings relative to pump and treat (groundwater) or excavation and landfill (soil and waste), and over 50% savings relative to Phytoremediation and Electrokinetic Remediation, with none of the field limitations. The actual costs will depend upon many specific aspects of the site including the metal of concern, the flow rates, the level of contamination, the physical set-up of the site, the geographical location, among others. As an example, a PIMS soil-mixing treatment of soil contaminated with 2000 ppm of Pb would cost between $20,000 and $30,000 per acre to a depth of 50 cm. Alternatively, a PIMS permeable reactive barrier could treat a groundwater, seep, or waste stream contaminated with 10 ppm Pb at 50 gpm for over 30 years for less than $100,000.
Adepoju, A.Y., P.F. Pratt, and S.V. Mattigod. 1986. Relationship between probable dominant phosphate compound in soil and phosphorus availability to plants. Plant and Soil, 92:47-54.
Altschuler, Z. S., 3.5. Berman, and F. Cuttita. 1967. Rare Earths in Phosphorites Geochemistry and Potential Recovery U.S. Geol. Survey Prof. Paper 575B, Denver.
Beckwith, R. 5.1964. Sorbed phosphate at standard supernatant concentration as an estimate of the phosphate needs of soils. AusL J. Exp. Agr. and An Hus. 5:52-58.
Bowhay, D. and the Interagency Fertilizer Testing Workgroup. 1997. Screening Surveys for Metals in Fertilizers and Industrial By-Product Fertilizers in Washington State, Washington State Department of Ecology Publication #97-341, Olympia, WA.
Chen, X.-B., 3. V. Wright, 3. L. Conca, and L. M. Peurrung. 1997a. Effects of pH on Heavy Metal Sorption on Mineral Apatite. Environmental Science and Technology , 31:624-631.
Chen, X.-B., 3. V. Wright, 3. L. Conca, and L. M. Peurrung. 1997b. Evaluation of Heavy Metal Remediation Using Mineral Apatite. Water, Air and Soil Pollution, 98:57-78.
Conca, 3. L. and 3. V. Wright. 1998. The UFA Method for Rapid, Direct Measurements of Unsaturated Soil Transport Properties. Australian J. of Soil Research, 36:291-315.
Davis, A., M. V. Ruby, and P. D. Bergstrom, 1992. Bioavailability of arsenic and lead in soils from the Butte, Montana, Mining District. Environmental Science and Technology, 26: 461-468.
Deer, F. R. S., R. A. Howie, and 3. Zussman. 1978. An Introduction to the Rock-Forming Minerals. Longman Group Ltd., London, 528 p.
Keto, L.S. and S.B. Jacobsen. 1987. Nd and Sr isotopic variations of Early Paleozoic oceans. Earth and Planetary Science Letters, 84:27-41.
Koeppenkastrop, D. and E.J. De Carlo. 1988. Adsorption of rare earth elements from seawater onto synthetic mineral phases. EOS Transactions of Amer. Geophysical Union, 69:1254.
Koeppenkastrop, D. and E.J. De Carlo. 1990. Sorption of rare earth elements from seawater onto synthetic mineral phases. Chem. GeoL, 95:251-263.
Kovach, 3. and R.W. Zartman. 1981. U-Th-Pb dating of conodonts. GeoL Soc. of Amer. Abstr. With Programs 13:285.
Ma, Q. Y., Traina, S. 3., and T. 3. Logan. 1993. In Situ Lead Immobilization by Apatite. Environ. Sci. Technol. 27:1803-1810.
McArthur, J.M.,1985. Francolite geochemistry --compositional controls on formation, diagenesis, metamorphism, and weathering. Geoch. Cosmoch. Acta, 49:23-35.
McArthur, 3.M., A.R. Sahami, M. Thirwall, P.3. Hamilton, and A.O. Osborn. 1990. Dating phosphogenesis with Sr isotopes. Geoch. Cosmoch. Acta, 54:1343-1352.
Misra, D. N., and R. L. Bowen. 1981 In: Adsorption from aqueous solutions, (ed. by P. H. Tewari), Plenum, New York.
Moody, T. E., and Judith Wright. 1995. Adsorption Isotherms: North Carolina Apatite Induced Precipitation of Lead, Zinc, Manganese and Cadmium from Bunker hill 4000 Soil, Technical Report BHI-00197, Bechtel Hanford, Inc., Richland, WA, 21 p.
Nriagu, 3.0. 1974. Lead orthophosphates, IV. Formation and stability in the environment. Geochim. Cosmochim. Acta, 38:887-898.
Ruby, M. V., Davis, A. and A. Nicholson. 1994. In Situ Formation of Lead Phosphates in soils as a Method to Immobilize Lead. Environ. Sci. Technol. 28: 646-654.
Shaw, H.F. and G.J. Wasserburg. 1985. Sm-Nd in marine carbonates and phosphates: implications for Nd isotopes in seawater and crustal ages. Geoch. et Cosmoch. Acta. 49:503-518.
Skinner, H. C. W. 1987. Bone: mineral and mineralization. In 3. A. Albright and R. Brand (eds.), The Scientific Basis of Orthopaedics, Appleton and Lange, Norfolk, CT.
Skinner, H. C. W. 1989. Low temperature carbonate phosphate materials or the carbonate-apatite problem. In Rex Crick (ed.), Origin, Evolution and ModeM Aspects of Biomineralization in Plants and Animals, Proceedings of the 5th International Symposium on Biomineralization, Arlington, TX, Plenum Press, NY.
Skinner, H. C. W. and C. W. Burnharn. 1968. Hydroxyapatite, Annual Report of the Director Geophysical Laboratory. Carnegie Inst., Washington, D. C.
Stanforth, R. and A. Chowdhury. 1994. In situ Stabilization of Lead-Contaminated Soil, Federal Environmental Restoration III and Waste Minimization II Conference Proceedings, New Orleans.
Wright, Judith. 1985. Rare Earth Element Distributions in Recent a,id Fossil Apatite: Implications for Paleoceanography and Stratigraphy. Ph.D. Dissertation, Geology Dept., Univ. Oregon, Eugene, Oregon.
Wright, Judith. 1 990a. Conodont apatite: structure and geochemistry, p.445-459. In Joseph Carter (ed.), Metazoan Biomineralization: Patterns, Processes and Evolutionary Trends, 28th International Geological Congress, Paleontological Society and American Geophysical Union, Washington, D.C.
Wright, Judith. 1990b. Conodont geochemistry, a key to the Paleozoic, p.277-305. In Willi Ziegler (ed.), 1st International Senckenberg Conference and 5th European Conodont Symposium (ECOS V) Contributions 111, Courier Forschungsinstitut Senckenberg, Frankfurt, Germany.
Wright, Judith, Richard S. Seymour, and Henry F. Shaw. 1984. REE and Nd isotopes in conodont apatite: variations with geological age and depositional environment, p. 325-340. In David L. Clark (ed.), Conodont Biofacies and Provincialism, Geol. Soc. Amer. Spec. Paper 196, Geol. Soc. Amer., Boulder, CO.
Wright, Judith, James F. Miller, and William T. Holser. 1 987a. Chemostratigraphy of conodonts across the Cambrian-Ordovician Boundary: western United States and southeast China, p.259- 286. In Ronald L. Austin (ed.), Conodonts: Investigative Techniques and Applications, Ellis Horwood, Ltd., London.
Wright, Judith, Hans Schrader, and W. T. Holser. 1987b. Paleoredox Variations in Ancient Oceans Recorded by Rare Earth Elements in Fossil Apatite. Geoch. Cosmoch. Acta, 51:631-644.
Wright, J., J L. Conca, J. Repetski, and J. Clark. 1990. Geochemistry and Microstructure of Conodonts from Jilin Province, China. In 1st International Senckenberg Conference and European Conodont Symposium Contributions III, Courier Forschungsinstitut Senckenberg, Frankfurt, Germany, 118:307-332.
Wright, J. V., L. M. Peurrung, T. E. Moody, J. L. Conca, X. Chen, P. P. Didzerekis and E. Wyse. 1995. In Situ Immobilization of Heavy metals it: Apatite Mineral Formulations, Technical Report to the Strategic Environmental Research and Development Program, Department of Defense, Pacific Northwest Laboratory, Richland, WA, 154 p
Xu, Y. and F. W. Schwartz. 1994. Lead immobilization by hydroxyapatite in aqueous solutions. J. Contaminant Hydrology, 15:187-206.